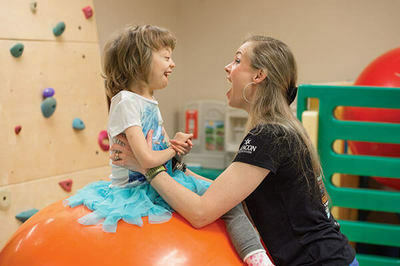
Perched on an orange ball taller than she is, Hope Kern giggles as her therapist tells her to stretch one of her long, thin arms and touch her image in a mirror.
“Come on, Hopey-Hope!” says Christy Bartek, a pediatric speech-language pathologist at the Children’s Therapy Center at Memorial Children’s Hospital in South Bend. “You can do it!”
Hope keeps speech and language pathologist Christy Bartek on the ball. Photo by Barbara Johnston
Flashing an impish grin, Hope, 7, reaches one red-polished nail toward the mirror but then bends the other direction and pulls off her pink sandal, tossing it onto the ground instead. She lets out a “yay!” and flops onto her back over the ball, laughing.
The exercises are meant to encourage Hope, who has Shprintzen-Goldberg syndrome (SGS), to engage muscles deep within her torso and hips, Bartek explains. The exercises might prevent Hope’s chest from developing mountains and valleys of unusual deformities, or help Hope develop better breath support. The truth is, Hope’s prognosis remains a mystery, because her disorder is so rare. A “rare disease,” by National Institutes of Health standards, is one that affects fewer than 200,000 people in the United States at any given time. Those diagnosed with SGS? Fewer than 50 in the world.
When a child is diagnosed with a disorder, any information is comforting. But Hope’s family hasn’t had that solace. Complete research has been performed on only about 300 of the 7,000 known diseases in the world, according to Kasturi Haldar, director of Notre Dame’s Boler-Parseghian Center for Rare and Neglected Diseases. SGS’s extreme rarity has kept it off a rare-disease list compiled by the National Organization of Rare Disorders (NORD), a nonprofit organization that provides one of three informational portals about rare diseases. Not until Notre Dame scientists from Boler-Parseghian met Hope and her family two years ago did any researcher have the time and resources to submit a detailed description of SGS to NORD, where it’s now under review.
Hope’s mother, Melissa Kern, contacted the Boler-Parseghian Center after the family moved from Bozeman, Montana, to South Bend in 2015, and after years of surgeries and extensive genetic testing finally proved that Hope, then 4, had SGS.
“It will be a huge, huge help to get this listed on NORD, to spread knowledge of this condition,” says her mother. “Right now, when you get a diagnosis, you get just a little packet that lists possible symptoms, and it’s so limited.”
Seen from behind, Hope looks like a skinny kid with maybe something wrong with her feet. Look at her face, and you’ll notice her wide-spaced, prominent eyes, small lower jaw and tapered, long fingers. Spend a few additional minutes, and you’ll hear that she responds more like a preschooler than a 7-year-old. You’ll also discover her charm. Hope has been welcomed as a jovial guest at the past two Rare Disease Day conferences in Jordan Hall each February. There, despite her disabilities, she skitters and skips between her parents and the poster displays, delighting anyone she meets with her exuberant, toothy smile.
“Hope,” says Joseph Farris, a biological sciences graduate student who is among those at Notre Dame researching SGS for the NORD database, “was very appropriately named.”
On June 4, 2010 — one day before Hope’s due date — Melissa and Tim Kern, along with some of their children, piled into the Bozeman Birth Center. They were eager to welcome Hope into the world, although they knew this baby, Melissa’s fifth, could be born with a disability. Months earlier, their midwife performed an ultrasound because she couldn’t locate Hope’s heartbeat with the standard fetal doppler probe. The ultrasound showed Hope was alive, but her umbilical cord had only one artery instead of two. This anomaly can sometimes indicate a genetic disorder. The midwife also couldn’t see Hope’s left foot, which didn’t appear in future ultrasounds, either.
Born at 8 pounds, 11 ounces, with a tuft of brown hair on her crown, Hope immediately cried like any other baby. Her parents were most delighted that she did, indeed, have two feet. But something about her was . . . off.
“When my midwife handed her to me, she was very floppy, like if you could imagine holding a big hunk of Jello in your arms,” Melissa says. Both she and Tim noticed that Hope was missing a nasal bridge — the raised portion of the nose usually found between the eyes. Her eyes were widely spaced and turned downward.
“My midwife kept saying I had a ‘very special little girl,’ and I was thinking she had Down syndrome,” says Melissa.
But the post-birth experience felt typical, she says, until Hope was moved out of the birthing center to a hospital and, later, whisked 200 miles away by helicopter to the Neonatal Intensive Care Unit at Community Medical Center in Missoula, not because of sudden changes in her status but because doctors couldn’t pinpoint her disorder.
“They started doing tests and wouldn’t let us touch her, and put her in a big bubble with monitors everywhere, and she really started going downhill after that,” Melissa says. “We were only allowed to touch her through the bubble, just like on her hand. It was very traumatic.”
Doctors ruled out Down syndrome. They considered several disorders during Hope’s first year, which included five surgeries (three for hernias) and weekly weight checks, because Hope’s weight fell sharply under average weight-gain curves. Tim rigged a gravity-feeding contraption from a microphone stand and a broken teapot handle so Melissa didn’t have to hold a bag of pumped breast milk for the 30 minutes it took Hope, who struggled to nurse or drink because of her underdeveloped lower jaw and high, wide palate, to finish a bottle.
By the end of Hope’s first year, a medical professional told the Kerns that Hope had a connective tissue disorder related to Marfan syndrome, “and she was naturally going to be thin,” Melissa says, lamenting that Hope had undergone an operation to place a gastronomy tube in her abdomen to deliver nutrients to her stomach — a surgery the Kerns now feel was unnecessary. The news, however, meant Hope was a step closer to being diagnosed.
Connective tissues are the glue that holds the body together. Cartilage, for instance. Even bone. Or the dura, a membrane under the skull that surrounds the brain. Faulty connective tissues create a cascade of problems throughout many of the body’s systems. In addition to skeletal abnormalities, those with Marfan syndrome and related disorders can suffer a fatal dilation of the aorta, the body’s largest artery.
Those life-threatening aortic aneurysms were what drove Dr. Hal Dietz, after four years as a pediatrician, into research in 1991. Now the director of the William S. Smilow Center for Marfan Syndrome Research at the Johns Hopkins University School of Medicine, Dietz was frustrated that he couldn’t cure children whose aortas were enlarged as part of their disease. By 2005, Dietz and researcher Dr. Bart Loeys described Loeys-Dietz syndrome, a Marfan-related disorder that Hope’s clinicians settled on after determining her aorta was dilated by age 2½. By 2008, Dietz and his colleagues identified a drug, losartan, that vastly slowed dilations — from about 3.5 millimeters per year to a half a millimeter.
After Hope’s Loeys-Dietz diagnosis, Melissa began combing a Facebook page dedicated to families of children with that disorder. Yet something still didn’t quite fit. Hope also has a moderate developmental disability, which wasn’t a symptom shared by Loeys-Dietz patients. Sensing the diagnosis was incorrect, Melissa reached out to Dietz, emailing him photos and a description of Hope’s symptoms.
Shprintzen-Goldberg syndrome was coined in 1982, but it wasn’t until 2012 that Dietz’s lab, simultaneously with another laboratory in France, homed in on the gene that causes the disorder. Loeys-Dietz syndrome and SGS have overlapping symptoms, including craniosynostosis, the premature fusion of the skull bones, which neurosurgeons corrected for Hope during a life-threatening surgery in 2015. SGS patients are less likely to have aortic malformations than patients with Loeys-Dietz, but when they do, the problem can be more severe. SGS patients also exhibit hernias, scoliosis, joint hypermobility and developmental delays.
Melissa’s email wasn’t an anomaly for Dietz. With the dearth of information about SGS, Dietz fields 40 to 50 calls or emails each week from patients or clinicians about that and other related syndromes.
“It is by the far the most rare of the connective tissue disorders,” Dietz says. “But I stress to people that knowledge is power, knowledge is hope, and I think this is a new beginning for patients, since identifying the gene.”
Genetic testing is expensive, and Hope’s medical disability insurance will pay for only three genes to test each year. Dietz, who wasn’t treating Hope, suggested to her doctors the exact genes to check that would determine whether Hope had SGS.
The results came back positive.
“To finally have a name attached with the disorder is viewed as a great victory and a first opportunity to find answers,” Dietz acknowledges.
Still, says Melissa, “The hard part has been finding information. There’s literally only scholarly papers out there.”
Those, plus Dietz and a Facebook page connecting a few families who have either received a clinical diagnosis or suspect their child has SGS. The information shared there proved life-saving for Hope. Another mother recommended to Melissa that a doctor check Hope’s upper two cervical vertebrae, to determine if they were being crushed by Hope’s skull. The doctor evaluated the two vertebrae, C1 and C2, and immediately scheduled Hope for surgery. She underwent a cranial-cervical fusion where one of her ribs was used to stabilize her neck.
“The literature doesn’t say to have this checked; it just says they can have a C1/C2 abnormality,” Melissa says, pointing to a paper containing a list of symptoms and noting that Hope could have died or become paralyzed. “You don’t even know how serious this can be.”
And this is where the work from Notre Dame’s Boler-Parseghian Center and NORD will bridge the information gap. Haldar, Farris and Barbara Calhoun, the center’s outreach coordinator, completed the paperwork in April to submit to the NORDdatabase. This portal of information on rare diseases is neither too technical nor too simple to be useful to patients. Haldar and Dietz tout it as the leading resource for the diseases it lists.
“Perhaps more importantly, it can provide a sense of community for patients and families, patient advocacy groups, clinicians and scientists with an investment in a rare disorder,” says Dietz, who will complete the final review of the Boler-Parseghian Center’s description before it’s published with NORD.
Hope has undergone 13 surgeries during her seven years. Tim and Melissa say they hope their daughter, whose aorta has shown little dilation since she started losartan last year, will have a surgery-free year. A surgery-free year isn’t only good for her (even though she appears to bounce back quickly from even the most nail-biting procedures, photographed with smiles the next day), but also the family budget. Medical bills are covered by Hope’s disability insurance, but other necessities — from travel, to special bicycles, to food, to special socks — are not. The family must purchase those items on Tim’s salary as a minister with Chi Alpha Campus Ministries or through the generous help of friends, family and service organizations.
Despite 13 surgeries in her seven years, Hope Kern’s exuberance has never waned. photo by Barbara Johnston
“We just have to be creative with what are truly needs and wants with the whole family,” Melissa says. “It’s a juggling act of balance and perspective with a whole lot of faith keeping it together.”
Meanwhile, other therapies continue. In addition to monthly visits with her pediatrician, Hope attends weekly speech therapy with Bartek and twice-weekly appointments at the Judd Leighton Speech and Language Clinic at Saint Mary’s College. At a recent appointment there, Hope played with baby dolls and pretended to cook plastic watermelons before pressing her face against the two-way mirror to smile at her dad, who she knew was watching.
The prolonged medical journey has been unyielding, Tim acknowledges. Still, Melissa tells anyone who asks, “Our family is bubble-wrapped in grace.”
They’re ready to tackle any future issues that arise and welcome the additional resources available once SGS is added to the NORD database.
“Just living close to Notre Dame has been a blessing, because they’re so passionate about rare diseases,” Melissa says. “I don’t know that we could get this close to getting Shprintzen-Goldberg on a national registry without it. It will be such a big help to parents.”